Advantages
- It works even if there is no known active compound.
- It provides the predicted structure with the binding score.
- The score can be divided into the contributions of the respective compound atoms.
Current Stage and Key Data
This technology has been applied to screen inhibitors of SARS-CoV-2 3CL protease and papain-like protease (see references).
Partnering Model
- Paid contract research conducted on behalf of pharmaceutical companies to screen ligands/inhibitors for target molecules.
- Handling the results of the contract research is negotiable.
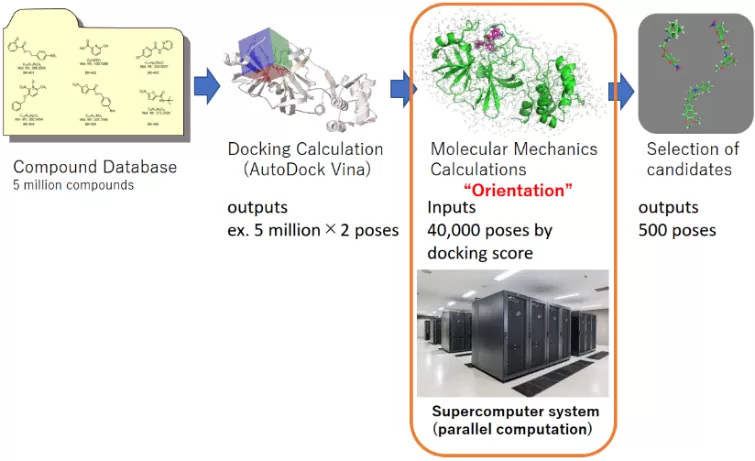
Background
When screening ligands/inhibitors for target molecules using AI analysis, it is difficult to find completely new hit molecules or different skeletons because it searches for compounds similar to existing active compounds or known compounds. The in silico screening of this technology can be performed even if there are no existing active compounds.
We have previously screened SARS-CoV-2 protease inhibitors and published papers on the results. We are also conducting collaborative research with many academic institutions both in Japan and overseas.
Principal Investigator
Tyuji Hoshino
(Laboratory of Molecular Design, Graduate School of Pharmaceutical Science, Chiba University)
Reference
Project ID: BK-04888