Advantages
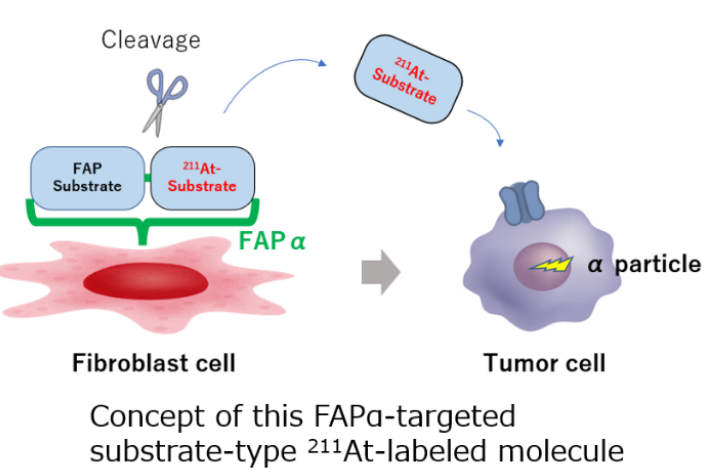
- Novel molecular design concept that does not regard FAPα as a binding target but rather exploits it as a cleaving enzyme
- This substrate‑type compound achieves high tumor uptake
- By attenuating the tumor stroma, this approach is expected to enhance intratumoral drug penetration and also alleviate the immunosuppressive state of the tumor microenvironment.
Current Stage and Key Data
Current Stage
- In vivo studies using pancreatic cancer and sarcoma mouse models that closely recapitulate clinical disease have demonstrated high tumor uptake and pronounced antitumor effects.
Key Data
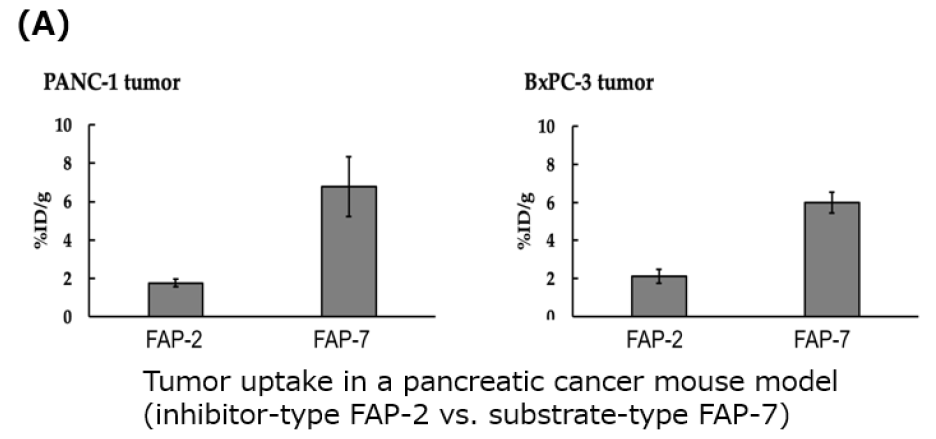
- In a pancreatic cancer mouse model, intravenous administration of the 211At‑labeled substrate compound (FAP‑7) resulted in an approximately three‑fold increase in tumor uptake compared with a conventional inhibitor‑type compound (FAP‑2).
- Furthermore, an optimized derivative of FAP‑7 showed tumor uptake exceeding 10%ID/g.
- In an osteosarcoma mouse model, the substrate‑type compounds exhibited marked tumor growth inhibition.
Background and Technology
In difficult‑to‑treat cancers such as pancreatic cancer and triple‑negative breast cancer, tumors are surrounded by a dense fibrotic stroma that acts as a physical barrier to drug penetration and is considered a major cause of treatment resistance and poor prognosis. In this fibrotic stroma, cancer‑associated fibroblasts highly and selectively express fibroblast activation protein‑alpha (FAPα), and FAPα‑targeted drug development has been pursued worldwide; however, the initial therapeutic outcomes haven't been satisfactory. Many of the FAP‑targeted agents currently in clinical development are inhibitor‑type (binding‑type) compounds, and their limited ability to reach tumor cells within the tumor mass is considered to restrict their therapeutic efficacy.
To overcome this limitation, research group designed a substrate‑type therapeutic that leverages FAPα as a cleaving enzyme rather than merely as a binding target. The compound consists of a FAP‑binding moiety linked to an alpha‑emitting radionuclide, astatine‑211 (211At), in a form that is efficiently taken up by cancer cells via transporters highly expressed in tumors. Once the substrate reaches the fibrotic tumor stroma, the peptide chain is cleaved by FAPα, releasing the 211At‑labeled amino acid, which is then selectively taken up into cancer cells. This mechanism enables dual targeting: disruption of the stromal barrier and direct alpha‑particle irradiation from within cancer cells, offering the potential for superior therapeutic efficacy compared with conventional inhibitor‑type FAP‑targeted agents.
Principal Investigator & Academic Institution
Invited Associate Professor. Yoshifumi SHIRAKAMI (Institute for Radiation Sciences, The University of Osaka)
Professor. Kazuko KANEDA (Institute for Radiation Sciences, The University of Osaka)
Patents
Patent (unpublished)
Partnering Model
The University of Osaka is seeking partnering opportunities with pharmaceutical and biotech companies active in radiopharmaceuticals and oncology.
We would be pleased to arrange scientific discussions with the principal investigators for interested parties. Various partnering schemes, including evaluation studies, collaborative research, and licensing agreements, can be flexibly discussed in line with your development strategy.